



医疗器械注册证信息怎么查询?
医疗器械注册证是依照法定程序,对拟上市销售、使用的医疗器械的安全性、有效性进行评价,决定同意其销售、使用后发放的证件,由国家食品药品监督管理总局统一制定。"


注册备案 · 临床试验 · 体系建立辅导 · 分类界定 · 申请创新
Recommend case

来源:医疗器械注册代办 发布日期:2026-01-04 阅读量:次
你们产品里的软件是不是隔一阵子就得升个级?可能是修复几个小漏洞,也可能是增加一两个用户想要的新功能。每次技术团队把新版本软件打包发过来,除了测试它稳不稳定,你脑子里是不是总会闪过一个念头:这个新软件,会不会影响咱们已经拿到的注册证?我是不是得去药监局办个什么手续?不办的话,将来会不会出问题?
")
这种担心太正常了。现在稍微复杂一点的二类医疗器械,不管是独立的软件,还是设备里内置的控制程序,基本上都离不开软件。软件一更新,事情就变得有点复杂。它不像换个螺丝钉那么简单,软件的变化看不见摸不着,但可能从根本上改变产品的行为。今天咱们就仔细聊聊,软件版本迭代到底会怎样触动你那张宝贵的注册证,让你心里有个明白账。
首先,你得扔掉一个天真的想法:软件更新是纯技术的事,跟注册没关系。
这个想法非常危险。从监管角度看,软件是你医疗器械产品不可分割的一部分。你注册证上批准的那个产品,是绑定着特定版本号的软件的。你把软件改了,就等于产品发生了变化。关键问题是,这个变化有多大?是大动干戈到需要重新审批,还是小修小补可以内部处理?
软件更新对注册证的影响,主要分三条路:需要走“注册变更”、需要走“变更备案”、或者啥也不用走(仅内部记录)。 走哪条路,不取决于你们程序员觉得改动大不大,而取决于这个改动对产品的“安全有效性”和“预期用途”有没有影响,有多大影响。
我们先说最麻烦的那种:你的软件更新,很可能需要走“注册变更”程序。
什么样的软件更新会掉进这个级别呢?但凡你的改动,影响了产品最初注册时确定的那些核心,基本就跑不了。
比如,你给软件增加了新的功能。原来你的医学影像软件只能看二维切片,现在你增加了三维重建功能。这相当于产品有了新的“预期用途”,风险也变了,你必须为这个新功能提供验证和确认证据,这通常就得申报变更。
比如,你修改了软件的核心算法。你的无创血糖仪,原来是基于A算法估算血糖值,现在你换成了全新的B算法。这直接关系到产品的“安全有效性”,是最核心的变更。你必须用临床数据证明新算法和原算法至少一样准、一样安全,这往往需要大量的工作,必然触发注册变更。
再比如,你的软件更新是为了适应硬件平台的重大更改。你要把设备从X86平台换到ARM平台,整个软件架构都移植了。这改动太大了,必须重新进行完整的验证和确认,注册变更也是免不了的。
走注册变更,意味着你要准备一套像样的申报资料,说明变更内容、理由,提交验证数据,等着审评老师审核批准。整个过程可能要好几个月,而且有通不过的风险。所以,对于这类更新,一定要提前规划,评估好了再做。
第二种情况,稍微简单点:走“变更备案”。
有些软件更新,确实有影响,但这个影响在监管看来是清晰、可控的,不需要惊动技术审评那么复杂的流程,备个案、告诉监管部门一声就行。
常见的例子是,你修复了一些不影响安全有效性的缺陷(BUG)。比如,用户界面某个按钮的颜色显示不对,或者报告打印的页眉页脚格式有误。这些纯属外观或非功能性问题,修复它们通常只需要进行充分的测试,证明修复后没引入新问题,然后就可以走备案程序了。
还有一种情况,你为了提升安全性和兼容性,进行的防御性更新。比如,更新了软件里的加密库,以应对新发现的网络安全漏洞;或者调整了软件,让它能兼容新版本的主流操作系统(如Windows 11)。这类更新是为了维持原有的安全有效性水平,而不是去改变它,通常也适用于备案路径。
备案比注册变更快得多,流程也简单,但同样需要你内部做好完整的验证记录,并正式向药监部门提交备案资料。
第三种情况,是最理想的:不需要走任何外部程序,自己内部记录好就行。
有些软件改动,实在太小了,小到对产品的任何方面都没有可察觉的影响。比如说,你改了几个代码注释,优化了一些不影响运行结果的日志输出信息,或者重构了部分代码但输入输出和行为与原来一模一样。
这类更新,你可以理解为“纯文字编辑”或“内部重构”。它们不需要惊动监管部门,但必须在你们公司的质量管理体系下做好记录。在你们的软件版本控制记录、设计开发文档里,要写清楚这次改了啥,为什么改,改完测试了没有,谁批准的。这些记录平时可能没人看,但万一哪天遇到体系核查或飞行检查,老师要看你们软件版本是怎么管理的,你得拿得出清清楚楚的台账。
那具体怎么判断一次更新该走哪条路呢?
你不能拍脑袋,得靠一个正经的流程:软件更新影响评估。每次计划更新软件前,都应该启动这个评估。
评估需要回答几个关键问题:这次更新修改了软件的哪些需求?是修改了已有功能,还是增加了新功能?这个修改,会影响产品的临床功能或性能指标吗?会影响产品的安全性吗(比如会不会死机、算错)?会影响用户如何使用产品吗?需要更新用户文档(比如说明书)吗?
根据评估结果,对照法规指导原则,就能基本确定变更的级别。这件事,需要研发人员、注册人员和质量人员坐在一起商量,单靠程序员是决定不了的。
这里有几个特别容易踩的坑,你千万留意:
第一个坑是偷偷改,不记录。觉得就是个小小补丁,随手就发布了,没走公司变更控制流程,也没留下任何正式记录。将来万一这个“小补丁”引发了一起不良事件,你在回溯调查时会极其被动,根本无法证明变更的合规性,可能要承担严重后果。
第二个坑是错误判断“轻微变更”。把一些实际上有影响的更新,自以为是地归到“内部记录”档。比如,你优化了一个计算速度,让结果输出快了0.1秒。你觉得这不算啥。但如果你的产品说明书里明确写了“测量时间:5秒”,你现在变成4.9秒,这就涉及性能指标的改变,可能就需要评估甚至备案了。
第三个坑是忽略了“滚雪球”效应。今天改一点,明天改一点,每次都觉得是微小变更,自己内部处理了。但一年下来,累计的改动量非常大,甚至产品面貌都变了。这时候,你其实已经脱离最初注册批准的状态很远了,但没有任何正式的变更记录,这会是一个巨大的合规隐患。定期回顾和评估软件的累计变更,非常重要。
实际操作建议
给你的软件版本定个清晰的命名规则,比如“主版本号.次版本号.修订号”。可以规定:修订号级别的改动,走内部记录;次版本号级别的,可能需要备案;主版本号升级的,很大概率要走注册变更。这能帮大家快速建立一个初步概念。
建立严格的软件变更控制流程。任何修改,必须提交申请,经过技术评估、注册法规评估、质量审核,批准后才能实施和发布。流程看起来繁琐,但它是你的安全阀。
如果你们公司对软件监管要求不熟悉,或者更新特别频繁、评估起来没把握,可以考虑引入外援。像思途CRO这样的专业机构,他们有专门的软件法规专家,能帮你搭建合规的软件生命周期管理流程,在具体更新时帮你准确分类,告诉你需要生成哪些证据。这能帮你省掉很多摸索和试错的成本,避免不小心踩到红线。
总之,软件版本迭代绝不是开发团队自己的事。它直接关系到你产品注册状态的合规性。把这件事纳入规范管理,每次更新前都问一句“这对注册证有什么影响”,养成这个习惯,能让你在产品的整个生命周期里都睡得更安稳。别等到出了问题,才回头去翻找当初到底改了啥,那可就太被动了。

站点声明
本网站所提供的信息仅供参考之用,并不代表本网赞同其观点,也不代表本网对其真实性负责。图片版权归原作者所有,如有侵权请联系我们,我们立刻删除。如有关于作品内容、版权或其它问题请于作品发表后的30日内与本站联系,本网将迅速给您回应并做相关处理。
思途医疗科技有限公司专注于医疗器械产品政策与法规规事务服务,提供产品注册备案申报代理、临床试验、体系建立辅导、分类界定、申请创新办理服务。

医疗器械注册证是依照法定程序,对拟上市销售、使用的医疗器械的安全性、有效性进行评价,决定同意其销售、使用后发放的证件,由国家食品药品监督管理总局统一制定。"
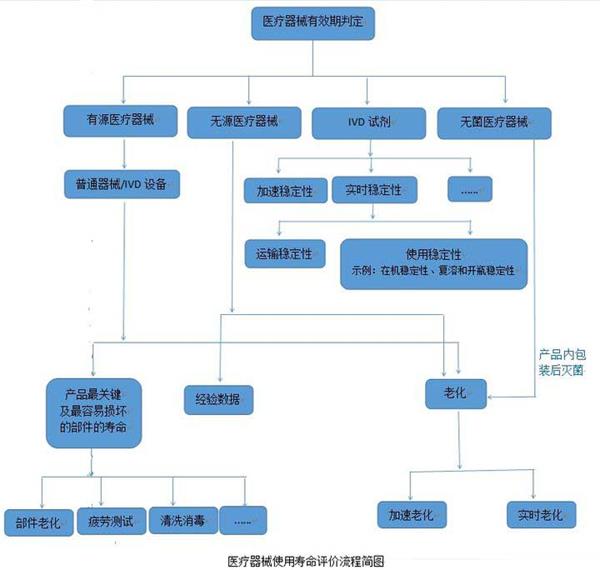
医疗器械的使用寿命是指医疗器械从规划、设计、生产、销售、安装调试到使用、维修、维护检测、报废的全过程。而医院使用的医疗器械的应用质量和安全管理在整个寿命过程中占重

从事医疗器械注册的小伙伴们可能都为同一个问题苦恼过,那就是医疗器械注册单元的划分。企业所设计开发出的产品,其所包含的产品范围,是否可通过一个注册单元完成注册,从而

随着医疗器械出口的日益增长,根据市场的需求各医疗器械生产厂商需要符合国家和地区的质量体系法规越来越多,所以经常会碰到出处于不同法规或标准的一些比较容易混淆的概念及

刚接触医疗器械CRO行业的小伙伴,在学习文件法规资料的同时,常看到一些英文类专业名词不知道是什么意思。下面,一起看看常见的医疗器械临床试验专业术语......"

目前,临床研究注册的要求是,前瞻性随机对照研究必须在研究开始前注册,观察性研究目前尚无统一要求,但有需要注册的趋势(脊柱外科前瞻性的研究不注册,文章一般很难发表,

在之前的文章中为大家简单介绍了国内医疗器械注册证的查询方法,很多读者表示非常实用,受益匪浅,但对于从事医疗器械研发工作的朋友们来说,他们希望获得更多的医疗器械信息

早在2017年11月24日CFDA和卫计委就联合发布《医疗器械临床试验机构条件和备案管理办法》(2017年第145号),把临床试验机构的资格认定,改为备案管理。目前,医疗器械临床试验机构备案

医疗器械注册检验报告的有效期,行业内常听到各种说法,一起来看一下效期是怎么规定的。"

任何国家医疗器械产品出口澳大利亚,需经历TGA注册,以下是关于澳大利亚医疗器械注册知识点,简单了解一下,文中大致概括了注册全流程,未标明细节,如有产品需要澳大利亚注册
六年
医疗器械服务经验
联系思途,免费获得专属《落地解决方案》及报价
咨询相关问题或咨询报价,可以直接与我们联系
思途CRO——医疗器械注册临床第三方平台
北京市 重庆市 河南省 安徽省 天津市 上海市 河北省 陕西省 山西省 内蒙古 辽宁省 吉林省 黑龙江省 江苏省 浙江省 福建省 江西省 山东省 湖北省 广东省 广西 海南省 四川省 贵州省 云南省

微信公众号
随时随地了解行业动态
客服二维码
询价咨询流程
扫码添加微信客服
填写您的需求,我们将尽快与您对接

专业CRO咨询服务提供商
专注于临床试验、GMP体系建立等器械研发全流程服务,多城市分公司就近对接
我们将在1个工作日内与您联系,信息仅用于业务沟通,严格保密