



山东省医疗器械工艺用水现场检查指南(鲁药监械〔2021〕35号)
为规范医疗器械现场检查,提升检查员现场检查能力,指导检查员对医疗器械生产企业工艺用水控制情况的检查,提高企业工艺用水质量安全保证水平,依据《医疗器械监督管理条例》


注册备案 · 临床试验 · 体系建立辅导 · 分类界定 · 申请创新
Recommend case

来源:医疗器械注册代办 发布日期:2024-01-15 阅读量:次
附件:医疗器械真实世界研究设计和统计分析注册审查指导原则(2024年第3号).doc
(图1)")
本指导原则旨在规范和合理引导真实世界数据在医疗器械临床评价中的应用,为申请人开展真实世界研究以及监管部门技术审评提供技术指导。
本指导原则是供注册申请人和技术审评人员使用的指导性文件,但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,需在遵循相关法规和强制性标准的前提下使用本指导原则。如果有能够满足相关法规要求的其他方法,也可以采用,但是需要提供详细的研究资料和验证资料。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则的相关内容也将进行适时调整。
本指导原则适用于医疗器械真实世界研究,不适用于按医疗器械管理的体外诊断试剂真实世界研究。本指导原则在《真实世界数据用于医疗器械临床评价技术指导原则(试行)》的基础上,结合目前积累的经验,进一步细化医疗器械真实世界研究设计和统计分析的一般要求。在当前发展阶段,真实世界证据在医疗器械临床评价中,主要作为已有临床证据的补充。
(一)试验性真实世界研究
实效性随机对照试验(pragmatic Randomized Controlled Trial, pRCT)是试验性真实世界研究的一种常见类型。
pRCT是指在真实或接近真实医疗环境下,采用随机、对照的设计比较临床实践中不同干预措施的治疗结果的研究,其主要目的是评估干预措施在常规临床实践中的效果。pRCT融合了随机化和真实世界数据优势,较好地控制了偏倚,其研究结果可为干预措施效果评价提供较高质量的真实世界证据。pRCT研究人群入选标准通常较广泛,人群代表性更好,但人群异质性通常较高,为保证统计分析具有足够的检验效能,pRCT所需样本量相对较大,适合需要生成在更广泛人群和临床情形中的临床证据的应用情形。例如,实效性随机对照试验可提供器械在不同亚组患者人群中的安全性、有效性数据,为器械受益风险评估提供更多有效信息。
(二)观察性真实世界研究
1.描述性研究设计
常见的描述性研究设计包括横断面设计、病例报告和病例系列设计等,不用于统计学上的因果推断。
在横断面设计中,全部测量在特定时点完成,主要用于描述接受了某种暴露因素(即:使用了待研究器械)的患者基本特征及健康状况、疾病恢复情况等分布情况,横断面设计可用于器械不良事件的描述性统计研究,为后续研究提供线索。病例报告设计用于描述临床一个或少数几个病例的详细临床特征,通常不描述事物的集中趋势或离散程度,病例系列设计是对多个病例资料进行归纳和总结,病例报告和病例系列设计提供对罕见或不寻常疾病/并发症的深入了解,特别是对于比较新的器械,其能够及时发现和记录其可能的副作用或并发症,同时为后续的更为严格和系统的研究提供线索和基础。
2.队列设计
在器械安全有效性评价中,队列研究是将某一特定人群按是否使用待研究器械以及使用不同器械分为不同的组别,追踪观察各组的结局发生的情况,比较各组间结局发生率差异,从而判定器械与结局之间有无关联及关联程度大小的一种观察性研究方法。队列设计根据研究时期的不同可分为前瞻性队列、回顾性队列、双向性队列。前瞻性队列研究中,根据预先制定的研究方案进行数据收集,可较好的控制数据质量。回顾性队列研究中,对过去已经产生的数据进行分析,数据质量可控性差,分析前建议先对数据的完整性和准确性进行确认。双向性队列在回顾性队列研究的基础上,继续前瞻性观察一段时间,其是将前瞻性队列研究与回顾性队列研究结合起来的一种设计,兼有上述两类的优点,一定程度上弥补了各自的不足。目前基于登记数据库开展的队列研究越来越多,例如利用CathPCI登记数据库比较Mynx血管闭合装置与其他同类已上市产品安全性事件发生率的回顾性队列研究,利用国家关节登记数据库比较不同制造商设计关节假体的翻修率等。
3.病例对照及其衍生设计
病例对照研究是以发生了结局事件的患者作为病例组,未发生结局事件的患者作为对照组,比较病例组与对照组使用待研究器械的比例,从而研究待研究器械与结局事件之间的关联性。回忆偏倚、对照组设置相关的选择偏倚、无法提供发病率等是病例对照设计的较为突出的限制。当待研究器械需观察的临床结局发生率较低时,采用实效性随机对照试验或队列设计所需样本量过大而不具有可行性时,可考虑采用病例对照设计。
病例对照衍生设计包括巢式病例对照和病例队列设计等,两种衍生设计兼具队列研究与病例对照研究的优点,在结局发生前收集暴露因素、混杂因素等信息;器械暴露组和对照组来源于同一人群,人群可比性更好;不需对队列全部人群进行测量。现阶段,基于登记数据库开展巢式病例对照设计越来越多。
4.病例交叉设计
病例交叉设计是一种常用于研究短期暴露与急性结局事件之间关系的观察性研究设计。在器械安全有效性评价中,每个病例在事件发生前后的不同时间段内充当自身对照,通过比较病例在事件发生前后对使用待研究器械情况来评估使用该器械与事件之间的关系。病例交叉设计可较好的控制不随时间变化的混杂变量,但也存在一些局限性,包括暴露效应期需较短,依赖患者回忆过去的暴露等。
(三)真实世界数据作为单臂试验外部对照
外部对照是指从其他试验或历史病例中找一组特征相似的研究对象作为对照组,真实世界数据作为单臂试验外部对照,是外部对照设计的其中一种。本指导原则不包含该类型设计具体的使用情形以及研究设计、统计分析等要求,相关内容另行制定指导原则。
(一)研究背景和目的
根据产品预期适用范围和产品技术特征,结合已有证据,在方案中阐明真实世界研究拟解决的安全有效性问题,明确研究目的。
(四)可行性评估
在研究目的确定后,申请人需评估是否具备开展真实世界研究的客观条件,主要考虑现有经验和知识积累是否足以事先确定影响临床结局的混杂变量,以及所需变量数据是否具有可获得性、数据量是否充分,数据质量是否满足需求。
首先,是否可事先确定影响临床结局的变量取决于现阶段对疾病、诊疗方法、器械临床相关知识和经验的积累程度,对于研究经验和知识积累还不充分的领域,申请人无法确保鉴别出对结局有重要影响的混杂变量,开展观察性真实世界研究时不能完全排除存在未测量或未调整混杂变量,研究结果的偏倚大小无法判定,结论稳健性难以保证。
其次,需评估变量数据是否可及和质量是否充分。在真实世界中,数据缺失较为常见。来源于真实世界的数据可能缺乏院外随访数据、结局指标(如功能评分、疼痛评分等)、影像学检查等。除此之外,还需考虑真实世界数据观察时间是否满足研究目的,研究人群是否具有代表性,前瞻性收集数据研究中样本量是否可保证足够的检验效能,现有数据质量是否可满足统计分析要求等。
(五)确定恰当的真实世界研究设计类型
申请人根据确定的研究目的,参考第二章内容,选择恰当的研究设计类型。如上所述,存在不同的真实世界研究设计类型,包括pRCT,队列研究、病例对照等,不同设计特点不同,适合应用的场景不同。
(六)研究流程图
考虑到不同类型真实世界研究实施过程存在差异,建议将实施过程以流程图的形式呈现,流程图按照时间顺序呈现研究过程中各具体事项(如伦理审查、人群筛选、接受干预措施、器械暴露后的数据收集相关事项(如检验、检查、评分量表填写等)以及各环节的质量控制措施等)。
(七)研究人群
研究方案需预先明确研究目标人群,目标人群定义清晰明确,避免歧义和模糊的表达,预先规定清晰的纳入和排除标准。
对于有多次器械使用记录的人群,需预先清晰规定纳入研究的标准,如在使用该器械前6个月无使用同类器械的记录,或将暴露定义为首次使用目标器械。预先核实不同临床机构是否采用相同的诊断标准,确保符合入选标准的患者均纳入研究。对于回顾性研究设计,需注意评估采用的数据源对预期适用人群的代表性。
(八)器械暴露
pRCT与传统RCT均采用随机方式决定器械暴露与否,但对于观察性真实世界研究,患者具体使用何种器械并非通过随机分组决定,而是在真实世界中根据实际情况(如医生偏好、患者病情)而定,存在选择偏倚风险。
对于回顾性真实世界数据,通常通过数据中使用器械的记录(如费用清单、手术记录)来判断患者具体使用了何种器械,需核实器械信息记录是否完整、准确,至少需包括器械制造商、型号规格信息。建议采用恰当的方法对器械暴露信息的准确性进行验证。
(九)对照组
实效性随机对照设计中,通过随机分组形成对照组。对于观察性真实世界设计,如队列研究、病例对照等,需根据研究目的以及设计类型,采用恰当的方式形成对照组,尽可能确保混杂变量在组间分布均衡是设置对照组的基本原则。根据研究目的,对照组可以是单一或多个制造商已上市同类产品,也可以是非器械类型的其他干预措施或安慰对照组。
队列研究设计中,依据研究目的可选择使用了其他同类已上市产品的患者构成对照组人群,也可选择未使用同类器械而使用了药品或其他诊疗方法的患者构成对照组人群。病例对照设计中,通常采用匹配的方法为病例组构建相应的对照组,巢式病例对照设计从同时期没有发生结局的研究对象抽样匹配形成对照组。病例队列设计在研究开始时从整个队列人群中随机抽样形成对照组,该对照组可重复用于不同结局研究,而巢式病例对照设计中对照组不能重复使用。
(十)评价指标
建议在研究方案中描述评价指标的选择依据和合理性,明确规定各评价指标的观察目的、定义、观察时间窗、指标类型、测定方法、计算公式(如适用)、判定标准(适用于定性指标和等级指标)等,并明确规定主要评价指标、次要评价指标和安全性评价指标。
对于回顾性真实世界研究,需注意确保不同临床机构对结局的定义相同,不漏记患者发生的结局事件。建议尽量选择客观指标,如死亡等,谨慎选择功能性评分等偏倚风险较大的主观性指标。
(十一)随访时间
需根据研究目的和设计明确随访时间的起始点及随访持续时间。对于植入性器械,通常为植入手术当天作为起始随访时间,对于多次治疗为一个完整疗程的器械,起始随访时间为最后一次治疗完成当天,注意治疗过程中的安全性事件也需观察。在回顾性真实世界研究中,研究者通常是通过病历、数据库或其他已有记录来收集数据,随访时间的长度和起始点可能受到现有数据可用性的限制。对于结局症状存在潜伏期或干预效果存在延迟的情形,可考虑在暴露和结局之间设置时间窗以防止逆因果混淆。
(十二)计算样本量和检验效能
对于回顾性真实世界研究,可基于可用的样本量估算检验效能。对于前瞻性真实世界研究,可基于预估的参数值计算样本量。不同研究设计估算样本量的方式不同,例如,横断面研究可基于预期达到的估计精度估算样本量,有对照组的研究设计基于组间比较差异、相对风险度、比值比等估算样本量。
对于因消除混杂偏倚涉及变量调整的真实世界研究,由于需预先估计的参数值较多,某些参数估算可能缺乏文献数据支持,样本量估算相较于传统随机对照临床试验复杂,需考虑的因素更多。例如,对于基于倾向性评分的分层调整统计需考虑层内效应值大小(如有效率、比值比、发生率等)、每层器械暴露组分配概率、倾向性评分重叠程度等。当模型过于复杂时,可以考虑使用基于模拟研究的方法帮助估计样本量。
(十三)质量控制
1.数据质量
(1)数据收集
建议制定完善的病例报告表和变量词典,依据病例报告表和变量词典收集和记录数据,并制定数据核查方案,确保数据无误。
前瞻性收集数据的研究设计中,建议事先规定具体的诊断、结局定义和判断标准,统一检验、检查和评分量表等评价项目的标准实施过程,确保收集数据的准确性和一致性。对于回顾性真实世界研究,需列明拟采用的数据源基本情况,包括所含的字段信息、患者数量、数据缺失、数据记录准确性等数据质量信息。方案中明确数据清洗的步骤与方法,若涉及多个数据库,方案中需明确链接的具体方法,以及数据链接准确性的验证方法。对使用到的各变量数据的准确性进行验证,采用算法自动提取数据时,对算法提取准确性进行验证。
(2)质量评价
见《真实世界数据用于医疗器械临床评价技术指导原则(试行)》第三章内容,建议对照该导则相关要求以表格形式逐项呈现数据质量评价结果,作为真实世界研究报告内容的一部分。
2.偏倚风险
偏倚是真实世界研究中需要特别关注和解决的问题,在真实世界研究设计、实施、分析和报告等各阶段均可能存在偏倚,真实世界研究方案需列出可能存在的偏倚、避免偏倚的策略以及在分析阶段处理偏倚的详细规定。对于观察性真实世界研究,可参考非随机干预性临床研究ROBINS-I评价工具对整体研究的偏倚风险进行评估。偏倚类型可以划分为选择偏倚、信息偏倚和混杂偏倚三大类,向下包括更多细分偏倚类型,不同研究存在的偏倚类型通常不同,部分仅列举部分真实世界研究常见的的偏倚类型,在实操中,申请人需根据实际情况具体分析研究中存在的各种偏倚风险。
(1)选择偏倚
① 研究人群纳入过程中产生的选择偏倚
确保对于不同组别始终采取相同的入选/排除标准,从相同的人群中筛选患者对于避免选择偏倚非常关键。建议按照时间顺序,连续不断地将符合入选/排除标准的研究对象纳入研究,以避免挑选患者,同时记录未纳入研究的病人及其原因。对于设置对照的研究,尤其是病例对照设计,需在设计中采取避免入院率偏倚的措施,如符合入选/排除标准的人群全部纳入,或当数据量过大时,病例组和对照组从同一人群中随机抽样确定。除此之外,还需注意自愿者偏倚等选择偏倚。
② 失访导致的选择偏倚
需在真实世界研究方案中尽可能设置充分的预防失访的措施,包括发生失访后可采用的补救措施,如通过额外的随访方式(如电话、登门拜访)弥补相关数据,与其他数据源(如医保数据、死亡登记数据等)链接等;
针对使用回顾性数据时可能存在的数据缺失情况,需在研究方案中预先明确数据缺失处理的方法和原则。针对缺失数据,需尽可能调查清楚失访的原因,若失访与干预措施或结局无关,可根据方案中预先规定的填补方法和原则进行填补。也可采用保守的方式进行填补,例如,器械暴露组填补为无效,对照组填补为有效。
(3)信息偏倚
① 干预措施偏离
在真实世界研究中,治疗中途由于各种原因干预措施可能发生偏离,例如患者主动要求更换治疗方式、医生改变治疗策略等,多次治疗的干预措施(如血液透析)或治疗时间长的干预措施(如呼吸机、体外膜肺氧合器),有更大可能出现干预措施偏离。在开展真实世界研究时,需提前考虑待研究器械出现此类偏倚风险的程度大小,制定措施减少干预措施偏倚的风险,同时全面、准确记录研究过程中的干预措施偏倚情况。
对于回顾性真实世界研究,在选择真实世界数据源时,需考虑该数据源是否详尽和准确地记录所用治疗方式及其治疗途中发生的变化。在临床实践中,还可能出现干预措施记录错误,如所用的器械制造商、型号规格记录错误,导致干预措施相关的信息偏倚,当怀疑存在记录错误的可能时,可考虑通过患者其他信息进行验证,如影像学下植入物形态、标记点特征、费用单上的价格等。
② 测量偏倚
测量工具、测量人员、测量方法和过程均可引入测量偏倚,优先考虑采取措施避免或减少测量偏倚,例如,制定详细的操作手册、培训工作人员、采用标准化的数据收集程序、核查数据质量、使用统一的方法收集、测量和解释信息;以下从三方面举例常见的减少测量偏倚的措施:
患者填写量表、回答问卷产生的测量偏倚:设置充分的培训,使患者能正确、一致的理解问题。
评价者来源的测量偏倚:对评价者施加盲法、选择客观的硬终点指标(如死亡等)、对评价者开展培训,不同评价者重复测量。
评价工具来源的测量偏倚:使用信效度经过验证的测量方法,使用精准的仪器。
对于回顾性真实世界研究,由于研究开始前数据测量工作已完成,以上适用于前瞻性真实世界研究的措施已无法实施,仅能对研究中涉及到的测量的准确性、一致性进行事后评估,若评估发现回顾性数据存在显著的测量偏倚,或无法评估回顾性数据的测量偏倚,建议谨慎使用该真实世界数据源开展研究。
③ 记录不准确
研究过程中,暴露信息、结局信息以及其他等任何信息记录的不准确或者错误都可能导致信息偏倚,应对此类信息偏倚,可考虑对数据记录者进行充分的培训,确保所有数据都按照统一的、预先定义的标准规范收集。定期进行数据审核和质量控制检查,确保数据的准确性和完整性。对于回顾性真实世界研究,需重点关注回顾性数据记录的准确性,对其开展评估和验证,确保数据记录具有足够的准确度后方可用于真实世界研究。
④ 回忆偏倚
尽量在设计阶段避免采用研究人群回忆的方式收集信息,尽量在数据产生时即记录至文档中。巢式病例对照设计可避免传统病例对照通过回忆获得暴露信息、基线数据等带来的回忆偏倚。
有些情况下,查看患者其他健康医疗资料可能有助于确认患者回忆是否准确。例如,如果患者回忆称自身接受干预措施后,有疼痛或发炎,可通过审阅该患者对应日期健康记录、服药记录、电子病历资料中是否存在相关信息,以进一步佐证。
⑤ 报告偏倚
选择性呈现有利的结果会造成选择报告偏倚,避免报告偏倚的最佳方法是在方案或统计分析计划中预先规定,建议将研究设计信息在公共网站(如中国临床试验注册中心、ClinicalTrials.gov、Open Science或ISPOR真实世界证据登记等)预先登记。
对于使用回顾性数据开展真实世界研究的情形,申请人需设置措施确保在正式统计分析前研究人员不可接触结局数据,避免研究人员在研究开始前为得到期望的统计结果开展数据挖掘行为。例如,在应用基于倾向性评分的统计分析方法时,可采取两阶段设计。第一阶段需构建结局数据防火墙、确定独立的统计人员、确定混杂变量、建立倾向性评分估计模型,直至第一阶段达到令人满意的混杂变量平衡后,再实施第二阶段的统计分析计划。
(4)混杂偏倚
混杂偏倚是指暴露因素与临床结局的相关(关联)程度受到其他因素的歪曲或干扰,使得呈现的研究变量与评价指标或结局变量的关系不是真实的,而是叠加了混杂效应的具有偏差的关系。
随机可同时控制已测量和未测量混杂因素,是控制混杂的最有力手段。pRCT以外的其他真实世界研究设计均不采用随机分组的方式,应用时需谨慎、全面评估潜在的各种混杂偏倚,并采取有效的措施尽可能的减少混杂偏倚。在设计阶段考虑使用限制、匹配和分层设计等其他方法控制混杂。在分析阶段,可应用分层分析、多变量回归分析、基于倾向性评分的调整方法等调整统计方法对混杂进行控制。值得注意的是以上方法均仅能控制已知且可测量的混杂。
如果所有混杂因素均已收集并正确建模,且样本量足够,理论上通过适当分析方法可消除混杂偏倚。然而在实践中难以获知全部混杂因素数据,部分混杂因素未知或不能测量,此部分偏倚称为未测量混杂偏倚。尽管可通过工具变量、E值(E-value)、敏感性分析等方法评估未测量混杂对结论的潜在影响,准确估计未测量混杂偏倚对结论的影响是一件困难的事情。
3.评估偏倚方向和大小
偏倚具有方向性,即低估或高估干预措施的效应值,偏倚亦有程度大小之分,相对较小的偏倚可能不会影响研究结论。在完成研究后,建议总结研究过程中仍然存在的偏倚,并评估对证据强度的影响。虽然偏倚评估并不是在任何情况下都具有可行性,仍然有一些方法可以帮助评估某些特定情形下的偏倚,例如对比失访研究人群特征与未失访研究人群特征,可能有助于评估失访导致的选择偏倚,比较不同临床机构对同一种评价方法的测量一致性可能有助于发现和评估测量偏倚。
(十四)伦理审查和知情同意
真实世界研究伦理审查和知情同意需符合研究当地法律法规要求,国内研究需符合《世界医学大会赫尔辛基宣言》和《涉及人的生命科学和医学研究伦理审查办法》等相关法规和指南的规定。
(一)统计分析计划
真实世界研究需要包括详细具体的统计分析计划,明确具体采用的统计方法和参数设定,以及统计方法和参数设定的理由和依据。真实世界研究更常涉及分层分析、回归分析、基于倾向性评分的调整性统计分析方法,相同的数据使用不同的分析方法,结果数值通常是不同的,若统计分析结果与设定的研究成功阈值/接受标准相近,可能存在采用不同统计方法研究结论不同的情形。即使使用相同的统计方法,参数选择等差异也可能导致结果不同。因此需尽可能详细具体提前规定统计分析计划,统计分析计划的详细程度需能确保按照计划执行分析时,不会存在可由分析人员自由选择分析方法和参数的情形。
(二)分析数据集
预先根据不同的分析目的定义不同的数据集,如有效性数据集和安全性数据集、亚组分析数据集等。
(十五)确定需调整的混杂变量
未采用随机分组的真实世界研究设计需预先确定需调整的混杂变量,需尽可能识别所有的混杂变量,以便在设计和统计分析阶段对混杂偏倚进行控制。通常可按照以下三条标准判断为混杂变量:(1)该变量与结局变量存在因果关系;(2)该变量与分组变量(暴露变量)存在关联;(3)该变量不是分组变量与结局变量因果路径中的中间变量。
建议首先制定合理的变量筛选流程,基于既往积累的专业知识和临床经验确定混杂变量,建议通过与临床专家团队和统计学专家团队进行讨论和确认。对于最终仍不能确认是否应纳入的变量,可对纳入和不纳入情形开展敏感性分析。列明变量纳入或不纳入调整的理由,提供支持性资料。在实际操作过程建议持保守的变量筛选态度,确定与治疗分配和结局变量均无关的变量才从模型中剔除,但也需注意避免纳入碰撞节点变量(Collider Variable)、工具变量(Instrumental Variable)、中间变量(Intermediate Variable),纳入变量时考虑是否存在多重共线性或交互作用。为清晰地展现各变量之间的因果关系,可使用有向无环图呈现(Directed acyclic graphs, DAG)。
(三)混杂调整统计分析
1.分层分析
分层分析是一种常用的控制混杂因素的方法,需在方案中预先规定层数的具体划分原则,明确具体使用的统计方法,如Mantel-Haenszel法,若使用其他加权统计方法,明确方法出处。
2.多变量回归分析
多变量线性回归分析需预先规定纳入的自变量,确定的混杂变量均需纳入到模型中去,当无法在设计阶段确定具体的自变量时,需在方案中明确具体的规则,以使纳入模型中的变量不可随意挑选。多变量回归分析中纳入的研究对象(及病例)的数量需满足模型中需要估计的参数所需的例数,存在一些经验帮助估计例数,例如EPV(events per variable)法。除此之外,需检验所使用模型的基本假设是否成立,如残差独立、残差期望为零、方差齐、分布假设、线性假设、Cox回归等比例风险假设等;恰当地处理多重共线性、交互作用;线性回归模型需具有可接受的拟合优度,需预先明确评估模型拟合优度的指标(多重决定系数、残差均方、马洛斯Cp统计量、赤池信息准则和贝叶斯信息准则等)、可接受阈值及其确定依据。
需预先明确多重共线性检验参数,如相关系数、方差膨胀因子、基于特征值的条件数等,预先明确判定是否存在多重共线性的阈值,以及阈值设定的依据,对于多重共线性的后续处理原则需有合理充分的论述。由于不能很好地探测比两两回归变量更复杂的多重共线性关系,不建议仅使用相关系数检验多重共线性。
是否纳入交互作用项需考虑专业知识经验和统计分析两方面。在专业知识和经验方面,如果已经存在先验信息显示回归变量间存在交互作用,或者专业知识可判断某变量与另一变量存在交互作用,需将该交互项纳入模型。若交互项存在统计学意义,但从专业无法判断交互作用是否真实存在,建议纳入和不纳入均开展统计分析,作为敏感性分析。
多变量回归模型中,优势比(odds ratio)和风险比(hazard ratio)等相对性统计指标不如率差和均值差等绝对性统计指标的临床意义直观,需注意对这些指标大小的临床意义解释。需预先在方案中明确判定研究假设成立的阈值。
3.基于倾向性评分的调整方法
基于倾向性评分的研究建议设计为两个独立的阶段,第一阶段主要内容包括识别混杂变量、估计样本量、构建结局数据防火墙、确定独立的统计人员、建立倾向性评分估计模型,过程中存在迭代,直至达到令人满意的协变量均衡为止,整个过程对结局数据保持盲态。第一阶段主要目的是实现器械暴露组和对照组组间均衡。第二阶段则是应用倾向性评分估计干预措施效应值。
在倾向性评分第一阶段(即寻找倾向性得分在组间均衡的阶段),部分特征人群由于未能在组间分布均衡而被剔除,此时需注意剔除部分人群后研究结论的外推性,需对过程有明确的记录,并对研究结论外推性有充分的论述。建议将研究器械组中的所有患者都包括在分析人群中,在倾向性评分未得到均衡的情况下,可增加对照组数据源。
估计效应值的方法包括分层、匹配、逆概率加权和回归等,需预先明确效应值估计采用的方法及相关参数。对于分层法,需预先明确分层具体划分,以及判定协变量组间均衡的阈值及其依据,层权重系数计算方式等;对于匹配法,需预先明确器械暴露组和对照组匹配比例(如1:1或1:n)、采用的匹配法(如精确匹配法、最近邻匹配法、卡尺匹配法等)、匹配成功判定阈值及其依据,通常单个患者数据仅用于1次匹配。
使用逆概率加权和回归法需注意解决以下问题。逆概率加权基于倾向性评分决定个体的权重系数,实现对效应值的加权,其在倾向性评分接近0或1的情形下权重会过大或过小,且逆概率加权对于倾向性得分模型准确性要求很高。回归法将倾向性评分直接纳入到模型中,假设结局变量与组别、倾向性评分的回归模型是正确的,这通常难以被证实。与逆概率加权相同,回归法对倾向性得分模型准确性较敏感,同时其在分析过程中由于涉及到模型优化而难以对结局数据保持盲态。
4.其他调整方法
用于控制混杂的调整方法还包括边际结构模型、工具变量和结构方程模型等较复杂的统计方法,目前在医疗器械临床评价中的实际应用较少。
(四)处理数据缺失
多种原因可导致数据缺失,如依从性差、缺乏改善、副作用、治疗体验差以及与研究无关的外部因素等,合理处理数据缺失十分重要。由于缺失机制无法通过已有数据进行判断,并且不同的处理方法可能会产生截然不同的结果,建议事先在方案或统计分析计划中明确处理方法,遵循保守原则对缺失数据处理方法进行规定。
应对缺失数据的最佳策略是通过合理的研究设计和高质量实施预防数据缺失。对于前瞻性真实世界研究,可通过筛选具有经验和负责的研究者、设置多种不同随访方式、采用阳性对照、采用易测量的结局指标、合理的数据收集表和快捷的数据录入方式、缩短研究时间、开展培训等方法减少数据缺失;对于回顾性真实世界研究,需评估数据库中数据缺失的程度,应报告每次测量的缺失和非缺失记录的数量。推荐使用敏感性分析(例如比较含缺失数据与不含缺失数据的患者的结果)来确定缺失的影响,若数据缺失较多,不建议使用该数据源开展真实世界研究。
目前常用的缺失数据填补法可分为单一填补和多重填补法两大类,需预先规定缺失数据具体的处理方法。单一填补法推荐使用保守的结转法,例如将器械暴露组缺失值均填补为无效,对照组缺失值均填补为有效;需注意单一填补法将减小方差,参数估计精度将被高估,体现为置信区间缩窄。对于多重填补,需预先规定填补模型、分析模型、插补次数、合并规则及相应确定依据。采用缺失信息比例、相对增加的方差、相对效率和参数稳定性等诊断指标对评估数据填补质量。在实际应用中,选择正确的填补模型非常关键,通常需要对数据有深入的理解以及对可能的缺失机制有合理的假设。
(五)亚组分析
若纳入研究的人群存在异质性,并且异质性可能导致不同效应值,需开展亚组分析。若不同亚组中效应值不一致,需注意此时研究结论对预期人群的外推性,避免产品在部分人群中具有显著的疗效,而在另外部分人群中无效。纳入\排除标准越宽,更容易出现异质性,可基于既往研究经验和知识预先在方案中确定亚组分析,未预先规定会降低亚组分析结果的可信度。
(六)敏感性分析
敏感性分析用于评估研究结果稳健性,在多种不同情形下均可能需开展敏感性分析,尤其对于观察性真实世界研究。真实世界研究中敏感性分析常见的应用情形包括违背模型假设、统计方法(如是否纳入交互项、共线性变量处理等)、变量选择、评估未被测量混杂的影响、评估干预偏离的影响、数据缺失填补、矛盾数据处理、离群值、变量定义不一致、不同人群亚组、基线不均衡等。需报告全部的敏感性分析结果,不能选择性报告。若敏感性分析的结果与主要分析的结果不一致,提示研究结论稳健性不够,可能需开展额外的研究进一步验证。
研究报告需遵循完整、准确、规范的总体原则。不同类型真实世界研究报告内容存在差异,pRCT研究报告内容可参考实效性试验CONSORT指南,队列设计、病例对照设计等观察性研究可参考STROBE指南,也可以参考其他适用的文件,如STaRT-RWE清单,以帮助提高临床报告要素的完整性。在以上的考虑基础上,需特别注意以下内容:
(一)研究设计:阐述选择该研究设计的原因,结合已有证据,阐明该研究拟回答的安全有效性问题。
(二)筛选流程图:提供研究对象的筛选流程图。对于回顾性真实世界研究,说明研究过程中如何从原始数据库中逐步筛选出合格的分析对象,给出原始数据库的样本量、每一个步骤排除的研究对象数目、相应的排除原因以及最后纳入分析的研究对象样本量。
(三)数据源基本特征描述:包括人群代表性、数据质量等;该数据源自身质控措施,基于该数据源发表的高水平文献等信息。
(四)人群基本特征描述:详细描述研究对象的基线特征,各组病例入选时的基线特征,基线数据是否平衡。
(五)数据准确性描述:对于回顾性真实世界研究,提供对使用到的各变量数据的准确性验证和/或评估结果,若采取自动方法从观察性数据库中提取变量数据,列明各变量具体的提取算法,提供相应算法的准确性验证数据。
(六)合并用药、合并治疗统计:记录和统计影响临床结局的其他干预措施(药物治疗及其他诊疗方法)实施情况。
(七)按照方案开展统计分析,呈现所有统计分析结果,包括:主分析结果、次要分析结果、安全性分析结果、亚组分析结果、期中分析结果、敏感性分析结果。
(八)数据缺失处理:描述缺失数据的数量及具体情况,列明缺失处理情况,与方案规定的符合性。若与方案规定的数据缺失处理方法不一致,提供合理的理由。
(九)矛盾数据处理:描述不同来源数据重要信息的统计量,针对矛盾数据的敏感性分析结果。
(十)偏倚风险及控制情况:以表格的形式列出可能存在的各细分偏倚类型,针对各偏倚逐个列出降低/消除偏倚的措施,并对应列出采取措施后残留的偏倚风险大小评估结果。
(十一)讨论和结论:讨论研究局限性,讨论潜在偏倚的方向和大小,分析剩余偏倚对临床证据强度的影响;讨论研究结果的外推性,例如考虑匹配人群和预期适用人群之间的差异,分析结果的外推性;结合研究目标、局限性、多种分析方法、相似研究的结果和其他相关证据,对结果进行谨慎、全面的解释。
[1]国家药品监督管理局.真实世界数据用于医疗器械临床评价技术指导原则(试行):国家药监局关于发布真实世界数据用于医疗器械临床评价技术指导原则(试行)的通告.2020年第77号[Z].
[2]高培, 王杨, 罗剑锋, 等. 基于真实世界数据评价治疗结局研究的统计分析技术规范[J]. 中国循证医学杂志. 2019;19(7):787-793.
[3]彭晓霞,舒啸尘,谭婧,等.基于真实世界数据评价治疗结局的观察性研究设计技术规范[J]. 中国循证医学杂志, 2019, 19 (7), 779–786.
[4]温泽淮, 李玲, 刘艳梅, 等. 实效性随机对照试验的技术规范. 中国循证医学杂志[J]. 2019;19(7):794-802.
[5]聂晓路, 彭晓霞. 使用常规收集卫生数据开展观察性研究的报告规范-RECORD规范. 中国循证医学杂志. 2017;17(4):475-487.
[6] Yue LQ, Campbell G, Lu N, Xu Y, Zuckerman B. Utilizing national and international registries to enhance pre-market medical device regulatory evaluation. Journal of Biopharmaceutical Statistics[J]. 2016;26(6):1136-1145.
[7] Sterne JA, Hernán MA, Reeves BC, et al. ROBINS-I: a tool for assessing risk of bias in non-randomised studies of interventions[J]. BMJ. 2016;355:i4919.
[8] Wang SV, Pinheiro S, Hua W, et al. STaRT-RWE: structured template for planning and reporting on the implementation of real world evidence studies[J]. BMJ. 2021;372:m4856.

站点声明
本网站所提供的信息仅供参考之用,并不代表本网赞同其观点,也不代表本网对其真实性负责。图片版权归原作者所有,如有侵权请联系我们,我们立刻删除。如有关于作品内容、版权或其它问题请于作品发表后的30日内与本站联系,本网将迅速给您回应并做相关处理。
思途医疗科技有限公司专注于医疗器械产品政策与法规规事务服务,提供产品注册备案申报代理、临床试验、体系建立辅导、分类界定、申请创新办理服务。
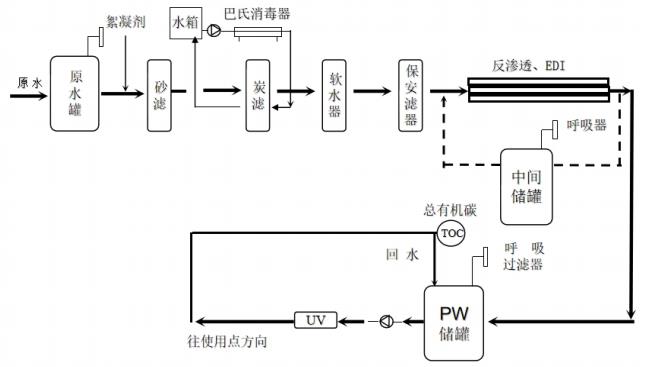
为规范医疗器械现场检查,提升检查员现场检查能力,指导检查员对医疗器械生产企业工艺用水控制情况的检查,提高企业工艺用水质量安全保证水平,依据《医疗器械监督管理条例》
《海南博鳌乐城国际医疗旅游先行区医疗机构制剂调剂使用管理暂行办法》已经3月24日海南博鳌乐城国际医疗旅游先行区领导小组会议审议通过,现印发给你们,请遵照执行。

为深入贯彻落实中共中央办公厅、国务院办公厅印发的《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号),按照国家药品监督管理局《关于扩大医疗器械

《海南博鳌乐城国际医疗旅游先行区临床急需进口药品带离先行区使用管理暂行办法》已经3月24日海南博鳌乐城国际医疗旅游先行区领导小组会议审议通过,现印发给你们,请遵照执行
为规范医疗器械注册人跨区域委托生产的监督管理,推进长江三角洲区域医疗器械跨区域监管,根据《中共中央办公厅国务院办公厅关于深化审评审批制度改革鼓励药品医疗器械创新的意见》《

2024年10月19日,在第 75 届世界医学会全体大会上正式通过了《赫尔辛基宣言》的最新修订版本;《赫尔辛基宣言》作为人类参与者的医学研究伦理

附件:血液透析用水处理设备注册审查指导原则(2024年修订版)(2024年第19号).doc血液透析用水处理设备注册审查指导原则(2024年修订版)本指导原则旨在指导注册申请人对血液透析用水处理设备注册申报资料的准备及撰写,同时也为技术审评部门提供参考。本指导原则是对血液透析用水处理设备的一般要求,注册申请人依据产品的具体特性确定其中内容是否适用。若不适用,要具体阐述理由及相应的科学依据,并依据产

附件:一次性使用医用冲洗器产品注册审查指导原则(2022年第41号).doc一次性使用医用冲洗器产品注册审查指导原则本指导原则旨在指导注册申请人对一次性使用医用

国家药监局关于发布免于进行临床评价医疗器械目录的通告(2025年第19号)发布时间:2025-05-13为做好医疗器械注册管理工作,根据《医疗器械注册与备案管理

附件:医疗器械监督管理条例(2025年修订版).doc医疗器械监督管理条例(2000年1月4日中华人民共和国国务院令第276号公布 2014年2月12日国务院第
六年
医疗器械服务经验
联系思途,免费获得专属《落地解决方案》及报价
咨询相关问题或咨询报价,可以直接与我们联系
思途CRO——医疗器械注册临床第三方平台
北京市 重庆市 河南省 安徽省 天津市 上海市 河北省 陕西省 山西省 内蒙古 辽宁省 吉林省 黑龙江省 江苏省 浙江省 福建省 江西省 山东省 湖北省 广东省 广西 海南省 四川省 贵州省 云南省

微信公众号
随时随地了解行业动态
客服二维码
询价咨询流程
扫码添加微信客服
填写您的需求,我们将尽快与您对接

专业CRO咨询服务提供商
专注于临床试验、GMP体系建立等器械研发全流程服务,多城市分公司就近对接
我们将在1个工作日内与您联系,信息仅用于业务沟通,严格保密